INTRODUCTION
Phenylketonuria (PKU) is an autosomal recessive genetic
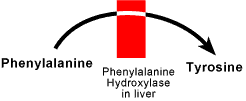
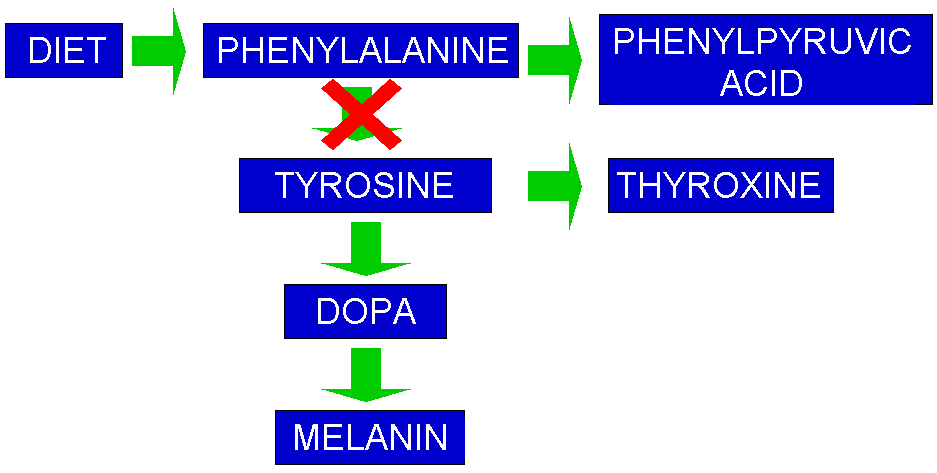
disorder where phenylalanine is not converted to tyrosine in the liver as
phenylalanine hydroxylase is absent. Excess phenylalanine accumulates in the
body and forms phenyl pyruvic acid, which damages central nervous system (CNS)
cells. Normal treatment for PKU involves limiting intake of Phenylalanine.
Before, PKU babies were immediately weaned from breastfeeding and fed with
phenylalanine-free formula and standard formula but after researchers discovered
that breast milk had lower levels of phenylalanine than standard formula, PKU
babies could alternatively be fed with breast milk and phenylalanine-free
formula.
However, fewer PKU
babies were breastfed than normal babies and PKU babies were weaned earlier
than normal babies.
As there are no studies about breastfeeding incidence and
period of PKU babies in America, thus the aim of this experiment is to find out
if breastfeeding is applicable for American PKU babies by comparing average
phenylalanine levels in breastfed and formula-fed PKU babies.
Methods:
This study was done in 2 hospitals with a total of 560 patient
beds. One of the hospital is a children’s hospital and is only pediatric
metabolic clinic program in the state. Hence, all infants diagnosed with PKU
are referred to. The retrospective chart review took place from 2006 to 2008.
Serial information on the Phe levels, infants’ medical, dietary and clinical
records that provided assessments of the infant from both dietitian and medical
perspectives provided the data. Using a medical record abstraction tool, data
were collected. The frequency of measuring phenylalanine levels is based on the
infant’s age and phenylalanine tolerance. This is done by using Guthrie cards
(specific filter paper onto which blood from a heel-prick is saturated).
Generally, phenylalanine levels will be assessed from every other day to twice
a week depending on phenylalanine level in blood. From 2002 till now, tandem
mass spectrometry is used to determine phenylalanine levels. Before that,
phenylalanine levels have been measured using an amino acid analyser. Being
more sensitive and specific, tandem mass spectrometry has improved the
detection of inborn errors of metabolism such as PKU.
RESULTS
5 levels of Phe levels were taken from each infant
(inclusive of the two newborn-screen results and confirmatory test, which were
elevated, thus skewing the data and hence were taken out of consideration) to
measure mean Phe levels for breastfed and formula- fed PKU infants. Results
show that more breastfed PKU babies (80%) than formulae-fed PKU babies (72%)
had a good mean Phe levels (120–360 μmol/L). This means that breastfed PKU
babies had a better chance of having desirable Phe levels. Formula-fed babies
had a higher chance of having too low mean Phe levels and the chances of having
too high Phe levels were the same for both groups. Infants with PKU living in
the central and coastal regions of Oregon have significantly higher mean Phe
levels.
Discussion:
Despite it being breast-fed or bottle-fed, most infants have
similar phenylalanine levels during the first year of life. Overall, more
breast-fed infants with PKU had phenylalanine levels within the normal range
(120–360 μmol/L). This supports previous research that breast-fed infants with
PKU can have phenylalaninelevels within the desired therapeutic range. We can
only hypothesize why infants with PKU living in the central and coastal regions
of Oregon have significantly higher mean Phe levels. Some possible causes are
no/little access to the tertiary clinic, lack of a professional support or
parental understanding for managing phenylalanine levels and the need to ensure
phenylalanine level remains normal. To clarify why infants diagnosed with PKU
have higher phenylalanine levels in the regions mentioned above, more research
have to be done.